Syndrome héréditaire qui associe un intervalle QT long à un risque élevé de syncope ou décès par arythmie ventriculaire (incidence 1 pour 2500-5.000 naissances soit environ 25/100.000). Il s’agit d’une « canalopathie » en rapport avec une mutation de l’un des gènes intervenant dans le fonctionnement de canaux ioniques membranaires des fibres cardiaques.
L’origine congénitale est secondaire à une anomalie génétique affectant des gènes variés, ce qui explique le caractère phénotypique variable (Ex. syndrome de Romano-Ward, Jervell et Lange-Nielsen, Andersen-Tawil, Timothy…) et une sensibilité voisine de 75% pour les tests génétiques (historique et état des connaissances sur les mutations génétiques en 2022 [3]). La transmission de ce syndrome est généralement de type autosomique dominant (risque de transmission de 50 % à chaque naissance [12].
Les risques rythmologiques sont les torsades de pointe qui se manifestent par des syncopes ou malaises à répétition, ou plus rarement une mort subite par fibrillation ventriculaire.
Clinique
Les présentations cliniques d’un syndrome du QT long congénital (SQTLC) sont variées et parfois trompeuses.
- L’expression clinique peut débuter dans l’enfance (SQTLC type 1) ou après la puberté (SQTLC type 2 ou 3).
- Le mécanisme déclenchant l’arythmie varie selon le type de SQTLC : voir infra.
- L’exercice physique, le stress émotionnel ou les stimulations sonores (SQTLC type 1 et 2) sont des facteurs déclenchants fréquents de ces complications parfois dramatiques. En règle générale, l’élévation du tonus adrénergique est un facteur arythmogène puissant chez ces patients par prolongation brutale du QTc ce qui explique le rôle préventif majeur des bêtabloquants [3].
- Des formes épileptoïdes peuvent être particulièrement trompeuses, ce qui justifie de faire un ECG et de mesurer précisément l’intervalle QTc chez les nouveaux patients épileptiques [9].
- L’expression ECG et la durée de l’intervalle QT varient dans le temps et avec l’âge [5]. Il est parfois normal lors de l’enregistrement ECG… Une déformation de l’onde T, de base ou après une ESV, doit rendre prudente l’interprétation d’un QT normal (voir ex [11]).
- La prise de certains médicaments peut parfois révéler une forme fruste de SQTLC (Cf. QTc drugs).
Mise au point française complète (mai 2021) [12]
Diagnostic ECG (ESC 2022 [4], AHA 2013 [5])
ECG 12 dérivations : examen clé, mais la mesure de l’intervalle QT peut être difficile, sources d’erreurs et de mauvaise prise en charge (cf. Intervalle QT) [2][8].
“Une valeur de QTc de durée normale (< 450-460 ms selon le sexe) est le témoin d’un risque très faible d’évènement cardiovasculaire”.
“Une valeur de QTc ≥ 480–500 ms sur des ECG répétés 12 dérivations est considérée comme diagnostic du SQTLC” [4].
“Une valeur de QTc ≥ 460–480 ms sur des ECG répétés 12 dérivations chez un patient présentant un épisode syncopal inexpliqué en l’absence de causes secondaires d’allongement de l’intervalle QT doit faire envisagé le diagnostic ” [4].
“Des QTc anormaux avec des valeurs < 480 ms (450-480 ms chez l’homme et 460-480 ms chez la femme) n’excluent pas le risque d’expression d’un syndrome du QTLC” (voir plus bas, score de Scharwtz).
Les ordinateurs peuvent fournir des valeurs fausses ou ininterprétables en raison de la fréquence cardiaque, de la méthode de calcul du QTc, de complexes QRS larges, d’irrégularités dans le rythme ou tout simplement de leur performance individuelle. Il faut donc valider manuellement toutes les valeurs qu’ils proposent !
La formule de Bazett est classiquement utilisée, mais d’autres formules comme celle de Fredericia (cf. Intervalle QTc) semblent plus adaptées quand la fréquence cardiaque s’éloigne trop de 60/min, en particulier dans les cas de bradycardie où le QTc “réel” est sous-estimé [7].
Le site https://www.qtcalculator.org/ est très utile. Il explique comment calculer un intervalle QT et permet de calculer la probabilité de syndrome du QTLC en fonction du genre, de l’âge et de la méthode employée pour le calcul du QTc (tangente ou threshold et formule) [10].
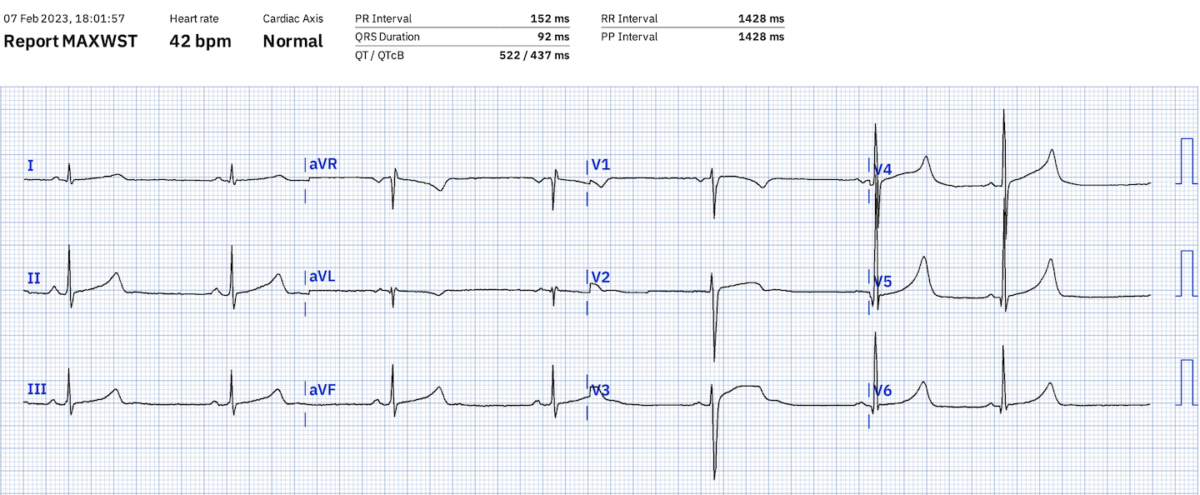
Légende. SQTLC1 (QTcF 489 ms). L’algorithme sous-estime la durée du QT (en fait 550 ms) et le QTc Bazett pour 42 bpm (QTcB 437 ms).
L’incertitude diagnostique est fréquente, si QTc « borderline » (440 < QTc < 480 ms)
Score de Scharwtz. Dans les cas de QT « borderline », une bradycardie sinusale, une morphologie anormale de l’onde T (onde T double bosse dans 3 dérivations), une alternance de l’onde T, une surdité, ou encore une syncope ou des torsades de pointes chez un sujet ayant des antécédents familiaux de QT long doit faire évoquer un SQTL (score de Schwartz) [1]. Un score > 3 ou une mutation génétique confirmée sont diagnostics du SQTC [4].
Génétique
Il existe au moins 17 sous-types de SQTLC avec mutations géniques identifiées [12]. Chaque sous-type possède des caractéristiques cliniques et ECG différentes (Goldenberg et Moss 2008 [1bis]). Les trois premiers sous-types représentent 90% des SQTLC (une dizaine connue en 2023).
- LQT1 – le plus fréquent – est caractérisé par une onde T à base élargie ou une apparition tardive d’une onde T normale (avec un sommet légèrement dévié à droite) (LQT1: typical broad-based T-wave pattern) ; Il expose des troubles du rythme survenant préférentiellement pendant un effort (notamment la natation). Il peut être caché au repos et ne se révéler qu’à l’effort en se manifestant par une mauvaise adaptation de la longueur du QT à l’élévation de la fréquence cardiaque, d’où une tendance particulière aux troubles du rythme dans ce contexte. C’est une présentation traîtresse qui fait qu’on ne peut pas écarter un SQTL devant un QTc normal sur un seul tracé (surtout au repos). La forme hétérozygote sans surdité s’appelle le syndrome de Romano-Ward). La forme homozygote de cette mutation donne le syndrome de Jervell et Lange-Nielsen avec une surdité congénitale et des troubles du rythme plus sévères.
- LQT2 est caractérisé par une onde T de faible amplitude et en double bosse (LQT2: typical bifid T-wave); Il provoque des troubles du rythme lors d’une émotion forte ou d’un bruit soudain (ou autre événement provoquant une libération abrupte de catécholamines). Ex. sonnette de téléphone, cris du bébé…)
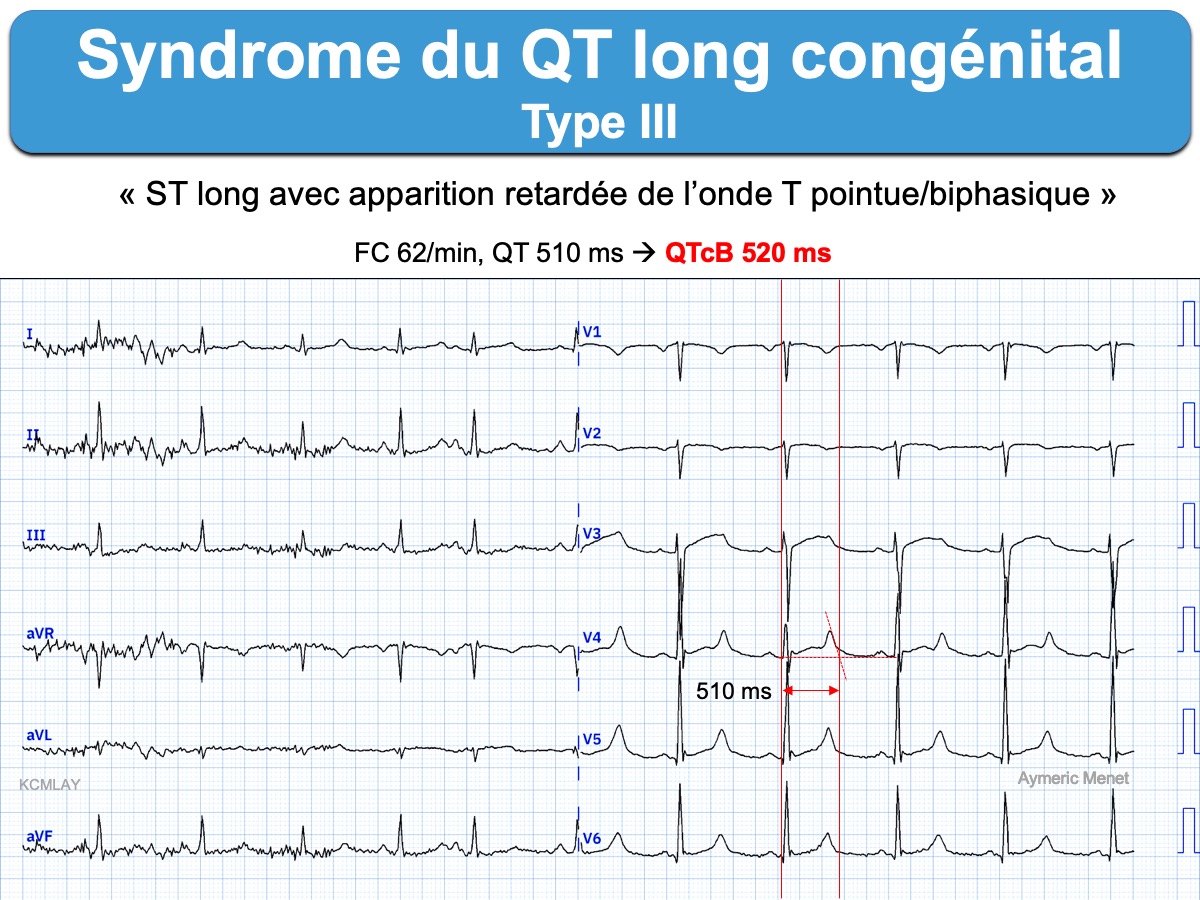
- LQT3 est caractérisé par un long segment ST et une apparition tardive de l’onde T qui peut être pointue ou diphasique (LQT3: typical late-onset peaked/biphasic T-wave). Il provoque des troubles du rythme au repos, en particulier pendant le sommeil. Le QTc a tendance à se raccourcir à l’effort (voire excessivement) et son pronostic est meilleur.
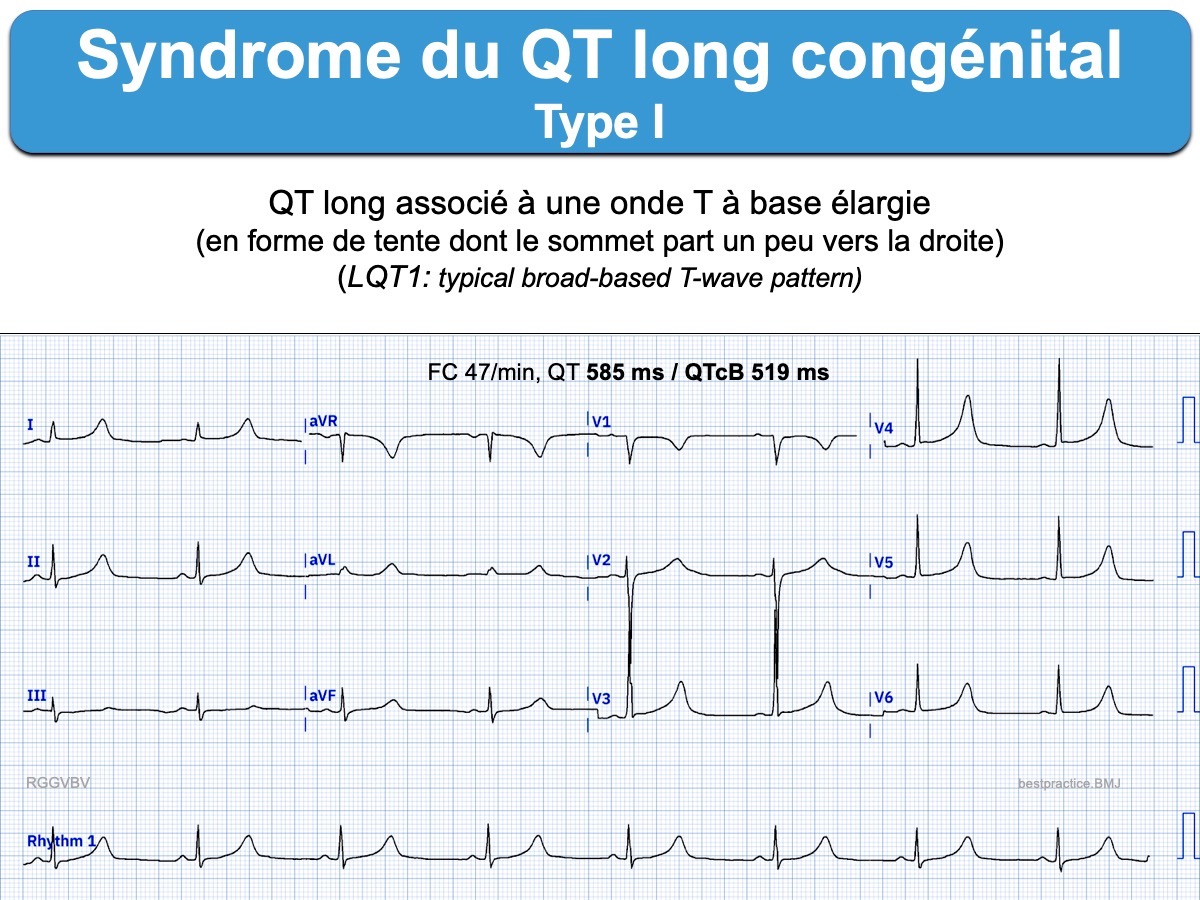
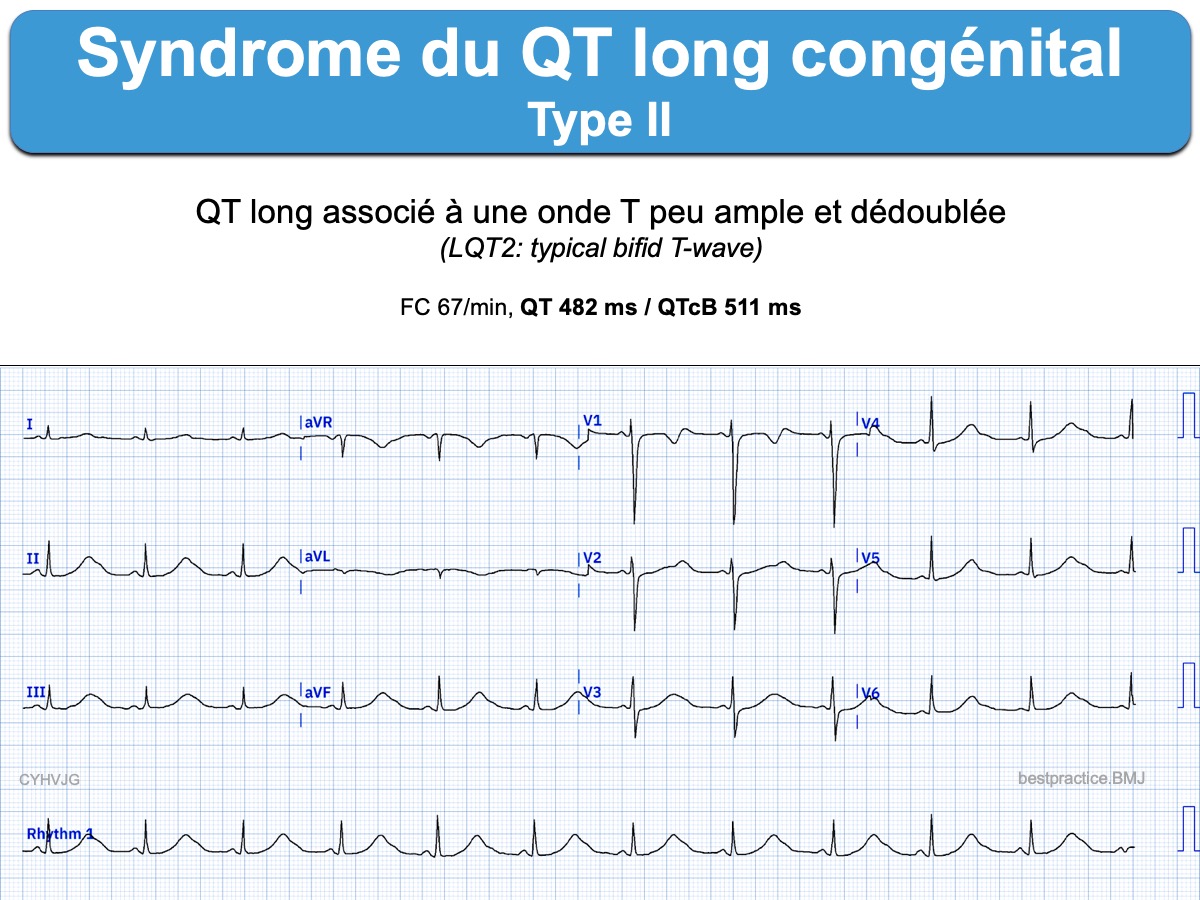
Un test par l’adrénaline peut être utile pour démasquer certains SQTL, en particulier le type I, lorsque le QT est normal à l’état de base.
Traitement (recommandations en cas de QT long [4][5] et en cas de TV [6]
Il varie selon les formes et repose sur une adaptation du style de vie, la contre-indication de nombreux médicaments, les bêtabloquants (classe I ou IIa selon la longueur du QTc) et le défibrillateur implantable chez les survivants d’une mort subite (classe I) avec syncope sous bêtabloquant (classe IIa) et les sujets à haut risque comme SQTL2, SQTL3, and QTc > 500 ms (classe IIb) [4][5][6].
Si vous souhaitez améliorer cette page, contactez-moi, merci
Vidéos YouTube
- Arythmie ventriculaire, Brugada, QT long
- Les syndromes du QT long : mise au point
Blog de SW Smith
- Do not trust the computerized QT when the QT is long
- Altered Mental Status, possible ingestion. What does the ECG show?Long QT Syndrome with Continuously Recurrent Polymorphic VT: Management
Références (réservées aux abonnés)
La suite est réservée aux membres et stagiaires du site.
Connexion | Devenir membre | Devenir stagiaire