Maladies du myocarde, associées à une dysfonction ventriculaire structurelle et fonctionnelle, en l’absence d’une maladie coronaire, hypertension artérielle, valvulopathie ou cardiopathie congénitale suffisante pour provoquer les anomalies myocardiques observées (Elliott P, ESC 2008 [2], Arbelo E, ESC 2023 [1]).
Classification
On distingue cinq catégories, d’origine familiale/génétique ou non (ESC 2008 [2]).
- Cardiomyopathie hypertrophique (CMH) : hypertrophie concentrique du VG ; une forme asymétrique touche essentiellement le septum et conduit à une sténose aortique sous-valvulaire dynamique [3]).
- Cardiomyopathie dilatée (CMD) : insuffisance systolique du myocarde, avec insuffisance cardiaque congestive, dilatation ventriculaire très importante, fraction d’éjection basse (FE < 0.3), et élévation des pressions de remplissage ; c’est la cardiomyopathie la plus fréquente (90% des cas).
- Cardiomyopathie arythmogène du VD : infiltration progressive du VD par du tissu fibreux et graisseux, accompagnée d’une dysfonction droite sévère et de troubles du rythme.
- Cardiomyopathies restrictives : insuffisance diastolique du myocarde, avec défaut de relaxation et de distensibilité, et élévation sévère des pressions de remplissage ; la fonction systolique est normale ; le ventricule gauche est de taille normale, mais l’oreillette gauche est dilatée ; c’est la forme la plus rare.
- Cardiomyopathies inclassables : la noncompaction du VG, le Barth syndrome… (familiale/génétique) et le takotsubo.
Parmi les CMH
: en majorité les causes familiales/génétiques, le cœur sportif (athletic training) et l’amylose cardiaque.
Parmi les CMD : les myocardites (toxique, infectieuse, immunitaire), la maladie de Kawasaki, l’hyperéosinophilie (Churg et Strauss’s syndrome), sarcoïdose, médicamenteuse (chimiothérapie), grossesse, endocrinienne (hypothyroidie…), carentielle (thiamine, carnitine, sélénium, phosphates, calcium), alcoolique, et la cardiomyopathie rythmique et les causes familiales/génétiques
Parmi les restrictives : les causes familiales/génétiques (Fabry-Anderson, hémochromatose…) et l’amylose, la sclérodermie, la fibrose endomyocardique (syndrome hyperéosinophilique, idiopathique, toxiques (sérotonine, méthysergide, ergotamine, agents mercuriels, busulfan), chimiothérapie (anthracyclines), le syndrome carcinoïde cardiaque, les cancers métastatiques, le cœur radique.
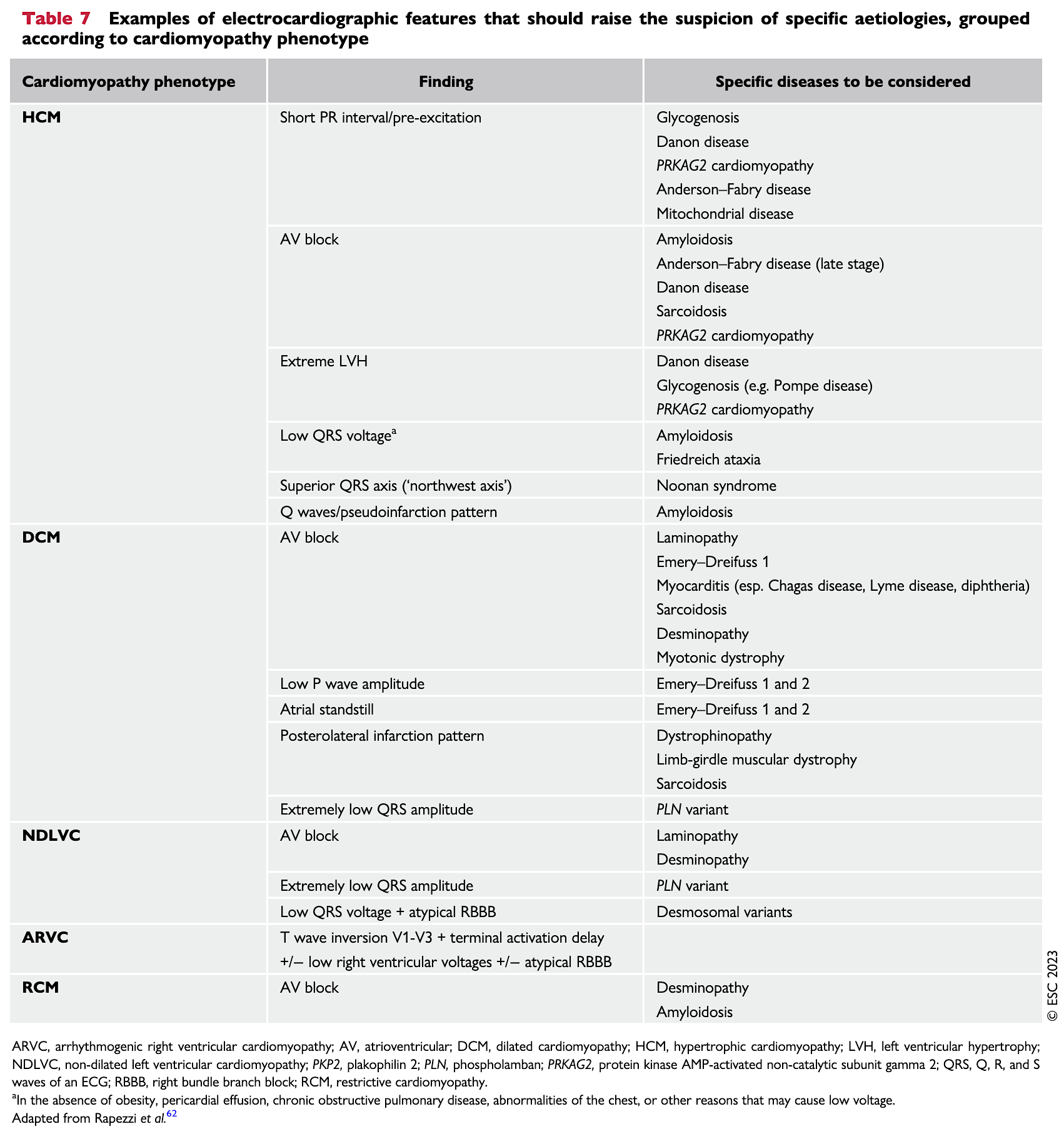
Diagnostic, prise en charge et traitement (ESC 2023) [1]
Voir imagerie cardiaque des cardiopathies
[1] Arbelo E, Protonotarios A, Gimeno JR, et al ; ESC Scientific Document Group. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-3626. (téléchargeable)
[2] Elliott P, Andersson B, Arbustini E, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008 Jan;29(2):270-6.
A cardiomyopathy is defined as ‘a myocardial disorder in which the heart muscle is structurally and functionally abnormal, in the absence of coronary artery disease (CAD), hypertension, valvular disease, and congenital heart disease (CHD) sufficient to cause the observed myocardial abnormality’
[3] Elliott PM, Anastasakis A, Borger MA, et al. 2014 ESC Guidelines on diagnosis and management of hypertrophic cardiomyopathy: the Task Force for the Diagnosis and Management of Hypertrophic Cardiomyopathy of the European Society of Cardiology (ESC). Eur Heart J. 2014 Oct 14;35(39):2733-79. –-> Cardiomyopathies are defined by structural and functional abnormalities of the ventricular myocardium that are unexplained by flow limiting coronary artery disease or abnormal loading conditions.
Lire aussi (pas de MEJ en 2022)
Maron BJ et al. Contemporary definitions and classification of the cardiomyopathies: an american heart association scientific statement from the council on clinical cardiology, heart failure and transplantation committee; quality of care and outcomes research and functional genomics and translational biology interdisciplinary working groups; and council on epidemiology and prevention. Circulation 2006; 113(14):1807-16. (téléchargeable)